Commercialising Pharmaceutical Technology
Commercialisation pathways of pharmaceutical technology can vary greatly between countries. This blog which will introduce you to the key player in Australia and broadly discuss some of the requirements and processes this regulatory body needs from applicants to take a drug from discovery to market.

This process mostly boils down to the regulatory bodies responsible for overseeing therapeutic goods available in their country. These regulatory bodies determine what criteria companies or sponsors need to meet in order to commercialise a therapeutic product, as well as what data and fees need to be provided. It is imperative for anyone looking to bring a therapeutic product into a new market to know precisely what the regulatory requirements are surrounding this process.
Therapeutic Goods Administration (TGA)
The TGA is a part of Australia’s Department of Health, which is responsible for the regulation of therapeutic goods. The TGA is responsible for the supply, import/export, manufacturing and advertisement of all therapeutic goods available in Australia. Through this single body, therapeutic goods are regulated via pre-market assessments, post-market monitoring, enforcement of industry standards, and ensuring all manufacturing facilities comply with the same standards whether they’re based in Australia or overseas.
The therapeutic goods that the TGA regulates in Australia fall under 3 main categories (source):
► Medicines: prescription, over-the-counter (OTC), and complementary/traditional medicines (vitamins, herbs, minerals etc).
► Biologicals: treatments made from or containing human cells/tissues.
► Medical devices: instruments (x-ray machines, hospital beds), implants, appliances (pacemakers)
The Australian Register of Therapeutic Goods (ARTG)
Every therapeutic good listed in the Australian Register of Therapeutic Goods (ARTG) can be supplied legally in Australia (source). This database is accessible through the TGA website for all members of the public. For every therapeutic good, the ARTG database contains the product name, formulation details, as well as the sponsor and manufacturer details. To be approved for registration of a product, the sponsor must complete and submit the relevant registration forms to the TGA (source). All forms are available through the TGA website, and the sponsor must determine which form is applicable to their product.
TGA’s Regulation of Prescription Medicines
The TGA uses an evidence-supported benefits vs. risks approach to the regulation of new prescription medicines. Their aim is to ensure the community can access new therapeutic advances within a reasonable timeframe from their discovery. As long as the benefits outweigh the risks favourably, the medicine will be registered for use in Australia.
To evaluate a new prescription medicine, the TGA assesses three types of data:
1. Chemical data describing the ingredients of the medicine
2. Nonclinical data from laboratories
3. Clinical data from trials
The chemical data is required for entry into the ARTG as well as for listing on product label claims. The nonclinical data indicates the organs that are most likely to be affected by the medicine, possible effects on reproductive organs and pregnancy, and the likelihood of the medicine being carcinogenic. The nonclinical data is important in identifying and assessing adverse reactions seen in clinical trials and potential further risks to the greater population that may not have been seen in clinical trials. The clinical data is gathered to assess both the efficacy of the medicine (ensuring it’s doing what the sponsor claims it’s intended to do) and the adverse events that occurred in a small population, and after exposure to the medicine for a specified period of time. Not all adverse effects from new medicines are captured in clinical trials due to the confined parameters of the trials (population size, time length, population diversity).
Based on their benefit vs. risk approach, the TGA understands that further safety concerns may emerge in other populations after the medicine has undergone clinical trials and been approved for market use. This is why the TGA continues to monitor all approved and registered medicines available on the market. If any new issues become known that were not seen in clinical trials, risk management plans (source), and a reporting system for adverse events (source) are utilised. The TGA monitors these reports and takes regulatory action if any concerns are flagged.
The TGA provides a resource to assist sponsors and applicants to register new prescription medicines or vary existing registrations in Australia (source). There are also similar sources for over-the-counter (OTC) medicines (source), and complementary medicines (source).
Clinical Trials in Australia
Clinical trials are a significant part of the regulatory process for new medicines wishing to enter the Australian market (source). Before beginning clinical trials in Australia, the sponsor must be aware of the requirements to import, export, manufacture and supply previously unapproved therapeutic goods for use in trials. There are two schemes that the TGA provides for sponsors to import and supply ‘unapproved’ goods for the use of clinical trials. These two schemes are not necessary to apply for if the sponsor is using goods in the clinical trial that are already listed in the ARTG database.
►The Clinical Trial Notification (CTN) scheme: is required for the trial sponsor to notify the TGA of the intent to sponsor a clinical trial involving an ‘unapproved’ therapeutic good.
►The Clinical Trial Approval (CTA) scheme: is required for sponsors seeking approval to supply ‘unapproved’ therapeutic goods in a clinical trial.
The TGA also provides a resource called the ‘Australian clinical trial handbook,’ which provides guidance on the legislative, regulatory and good clinical practice (GCP) requirements for clinical trials in Australia using ‘unapproved’ medicines (source). This handbook has information relevant to the roles of trial sponsors, ethics committees, investigators and approving authorities.
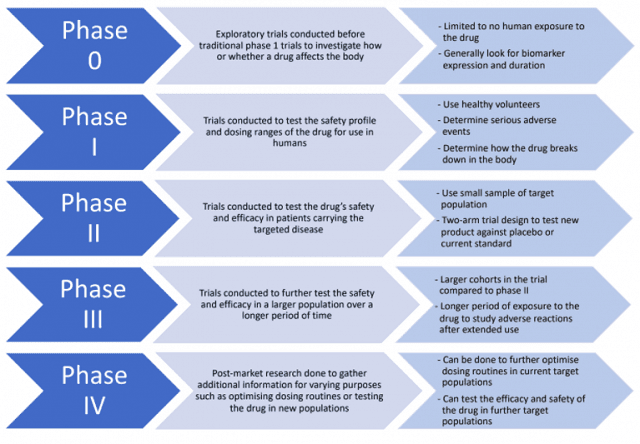
The clinical trial roadmap, through all five phases (phases 0-IV) is not necessarily set in stone. Depending on the ingredients of the drug, or the extent of the data (nonclinical and clinical) already available for the drug, some phases may be skipped or combined. For example, if trials have been run in other countries, the ethics committee reviews the trial design and the gathered data and will deem them adequate. In that case, the sponsor may be able to skip phases 0/I and begin at phases II/III. If the sponsor already had clinical trial data from overseas, they are advised to contact the TGA to discuss whether they are eligible to move ahead in the trial roadmap.
Conclusion
If you are planning on introducing and registering a new product in a market you are unfamiliar with, it is important to first identify the regulatory body you must work with and familiarise yourself with their processes and requirements. It is also worth seeking help from knowledgeable professionals in the market or region you are considering. Consultants specialising in commercialisation practices can also assist you in multiple ways including ensuring that all provided data is necessary, complete application processes, and provide market insights. By doing so, you can ensure that your entry pathway avoids as many speed bumps as possible, and allows for a smooth transition into new regions.
Disclaimer: The information provided in this blog is strictly for educational purposes to explain the regulatory environments of therapeutic goods in different jurisdictions. It does not constitute investment, accounting, financial, legal, or tax advice. It has been prepared without taking into account your personal objectives, financial situation or needs. Before acting on any information you should consider the appropriateness of the information having regard to your objectives, financial situation and needs.




